
Works
Talks
Timeline
CV
Me
(???)
Fabrizio Frasca
Hey! I am Fabrizio. I am a Postdoctoral Fellow at Technion, working with Prof. Haggai Maron on Geometric Deep Learning.
My interests revolve around principled and effective learning over structured data, with particular emphasis on the roles of Equivariance and Expressiveness. Recently, I’ve been working on architectures to learn from (structured) computational traces of Large Language Models (LLMs) for the automated detection of problematic behavioral patterns such as hallucinations. I also extensively work on methods for learning on graphs, with a focus on designing efficient and expressive Graph Neural Networks (GNNs).
Speaking of graphs, I am also one of the co-founders and co-organisers of the GLOW (Graph Learning on Wednesdays) reading group.
Previously, I have obtained a PhD in Computing from Imperial College London under the supervision of Prof. Michael Bronstein, with my research focussing on overcoming the intrinsic representational limits of GNNs. I have explored extensions of message-passing schemes that can capture non-trivial meso-scale topological patterns in networks, and equivariance to symmetries as an overarching design principle to design provably expressive architectures.
In the past, I have conducted more applied research on Machine Learning approaches for problems in the realm of Computational Biology and Bioinformatics, specifically, drug repurposing and epigenetic gene expression regulation.
I have also been a Machine Learning Researcher at Twitter Cortex from 2019 – acquisition of Fabula AI – to early 2023.
News
[2025/11] Really glad to share our LOGML project 'Lost in Serialization: Invariance and Generalization of LLM Graph Reasoners' will be presented at the AAAI 2026 workshop on 'Graphs and more Complex Structures For Learning and Reasoning'!
[2025/11] News from AAAI 2026: our 'Beyond Next Token Probabilities' has been accepted as a poster in the main conference track!
[2025/10] Extremely glad I have been selected, even this year, amongst NeurIPS' best reviewers!
[2025/9] Our paper 'Beyond Token Probes: Hallucination Detection via Activation Tensors with ACT-ViT' has been accepted to NeurIPS 2025! See you in San Diego :)
[2025/9] New preprint out: 'Neural Message-Passing on Attention Graphs for Hallucination Detection': we cast hallucination detection in LLMs as a learning task over the heterogeneous, directed graph determined by LLMs' computational traces, including attention maps and activations.
... see more here ...
Selected Works
(* indicates equal contribution; see here for a complete list).
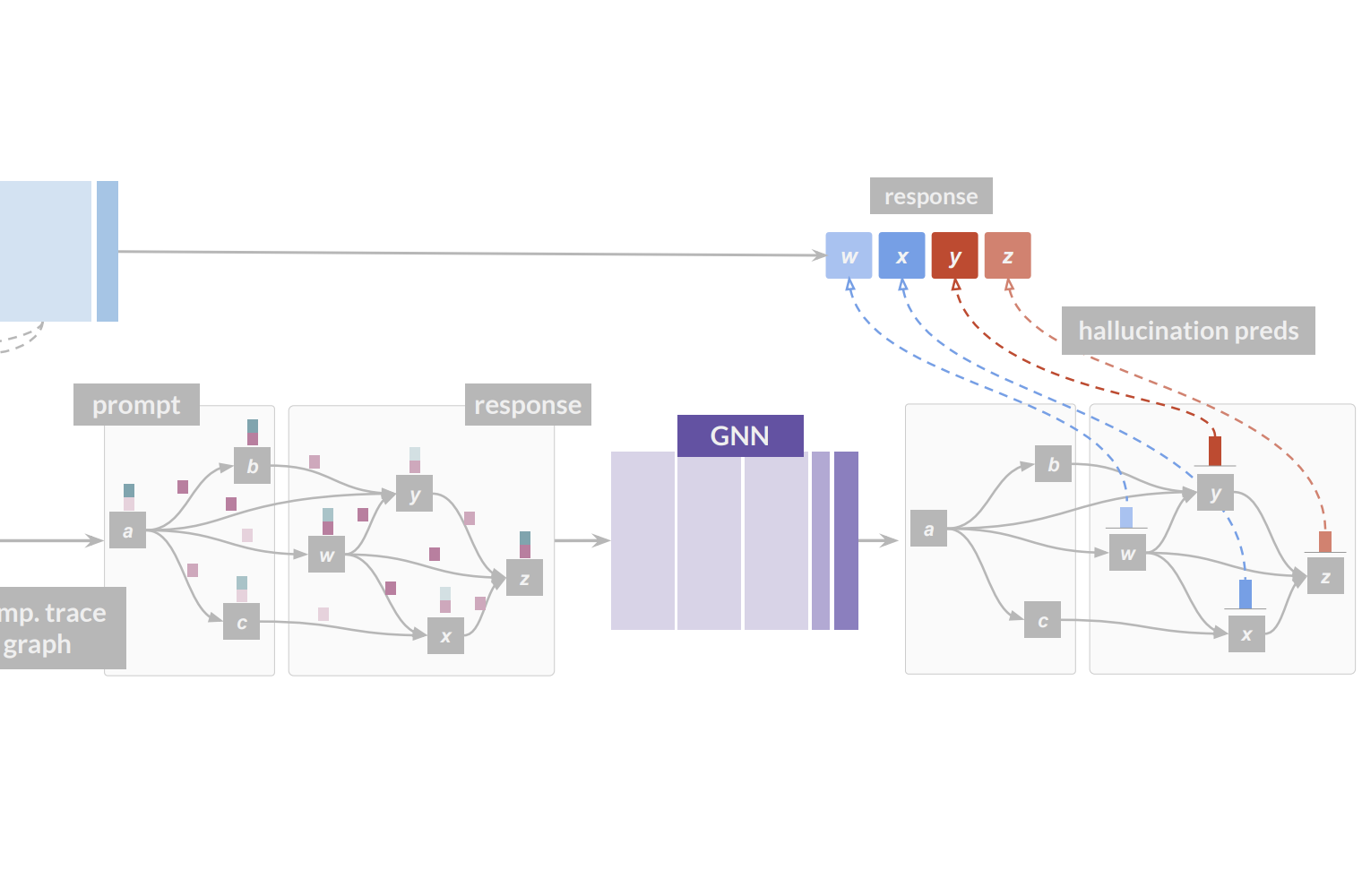
Neural Message-Passing on Attention Graphs for Hallucination Detection
Fabrizio Frasca*, Guy Bar-Shalom*, Yftah Ziser, Haggai Maron
(paper)
Abstract
Large Language Models (LLMs) often generate incorrect or unsupported content, known as hallucinations. Existing detection methods rely on heuristics or simple models over isolated computational traces such as activations, or attention maps. We unify these signals by representing them as attributed graphs, where tokens are nodes, edges follow attentional flows, and both carry features from attention scores and activations. Our approach, CHARM, casts hallucination detection as a graph learning task and tackles it by applying GNNs over the above attributed graphs. We show that CHARM provably subsumes prior attention-based heuristics and, experimentally, it consistently outperforms other leading approaches across diverse benchmarks. Our results shed light on the relevant role played by the graph structure and on the benefits of combining computational traces, whilst showing CHARM exhibits promising zero-shot performance on cross-dataset transfer.
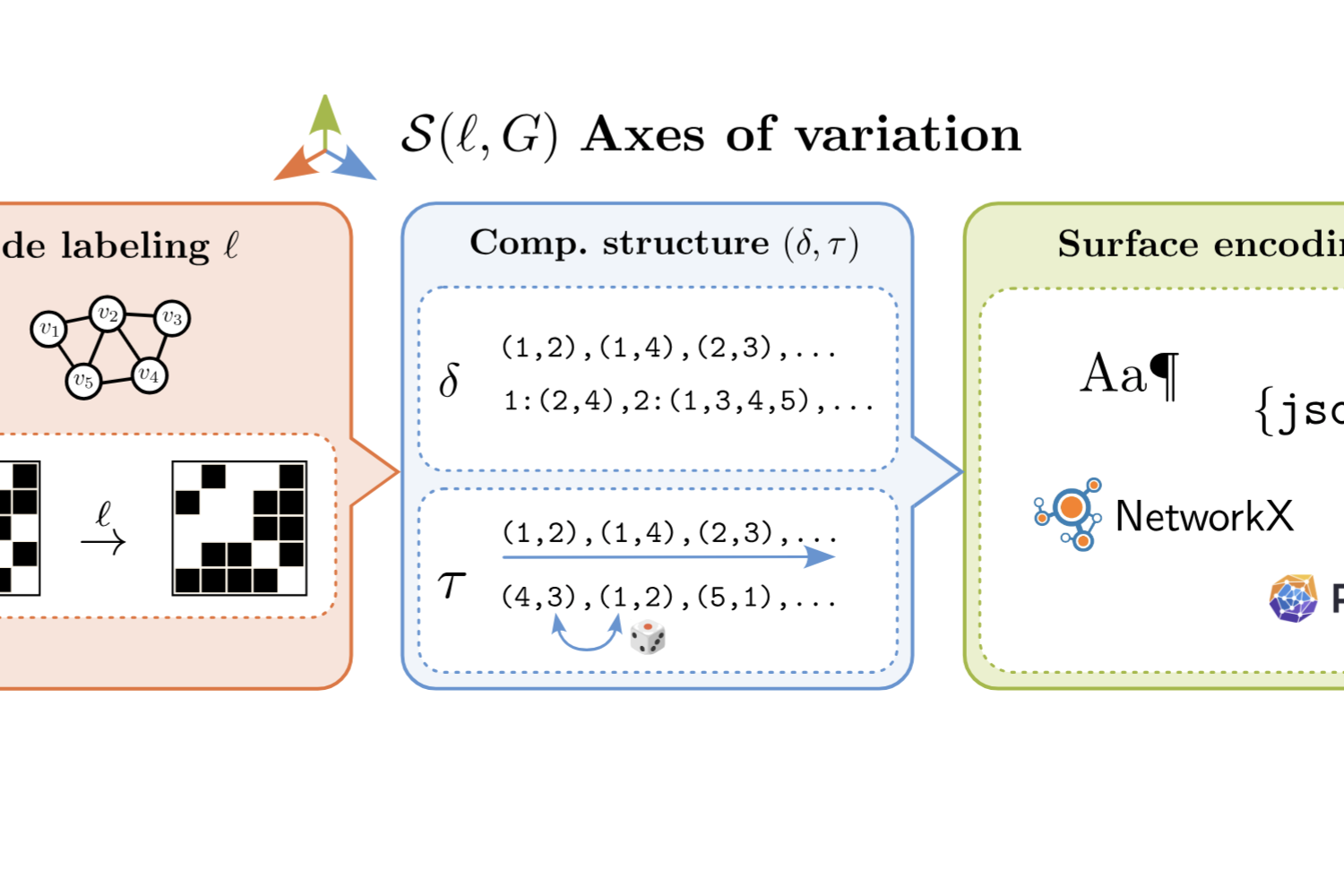
Lost in Serialization: Invariance and Generalization of LLM Graph Reasoners
Daniel Herbst, Lea Karbevska, Divyanshu Kumar, Akanksha Ahuja, Fatemeh Gholamzadeh Nasrabadi, Fabrizio Frasca
(paper)
AAAI 2026's workshop on Graphs and more Complex Structures for Learning and Reasoning
Abstract
While promising, graph reasoners based on Large Language Models (LLMs) lack built-in invariance to symmetries in graph representations. Operating on sequential graph serializations, LLMs can produce different outputs under node reindexing, edge reordering, or formatting changes, raising robustness concerns. We systemati- cally analyze these effects, studying how fine-tuning impacts encoding sensitivity as well generalization on unseen tasks. We propose a principled decomposition of graph serializations into node labeling, computational structure, and surface encoding, and evaluate LLM robustness to variations of each of these factors on a comprehensive benchmarking suite. We also contribute a novel set of spectral tasks to further assess generalization abilities of fine-tuned reasoners. Results show that larger (non-fine-tuned) models are more robust, and fine-tuning reduces sensitivity to node relabeling but may increase it to variations in structure and format, while it does not consistently improve performance on unseen tasks.
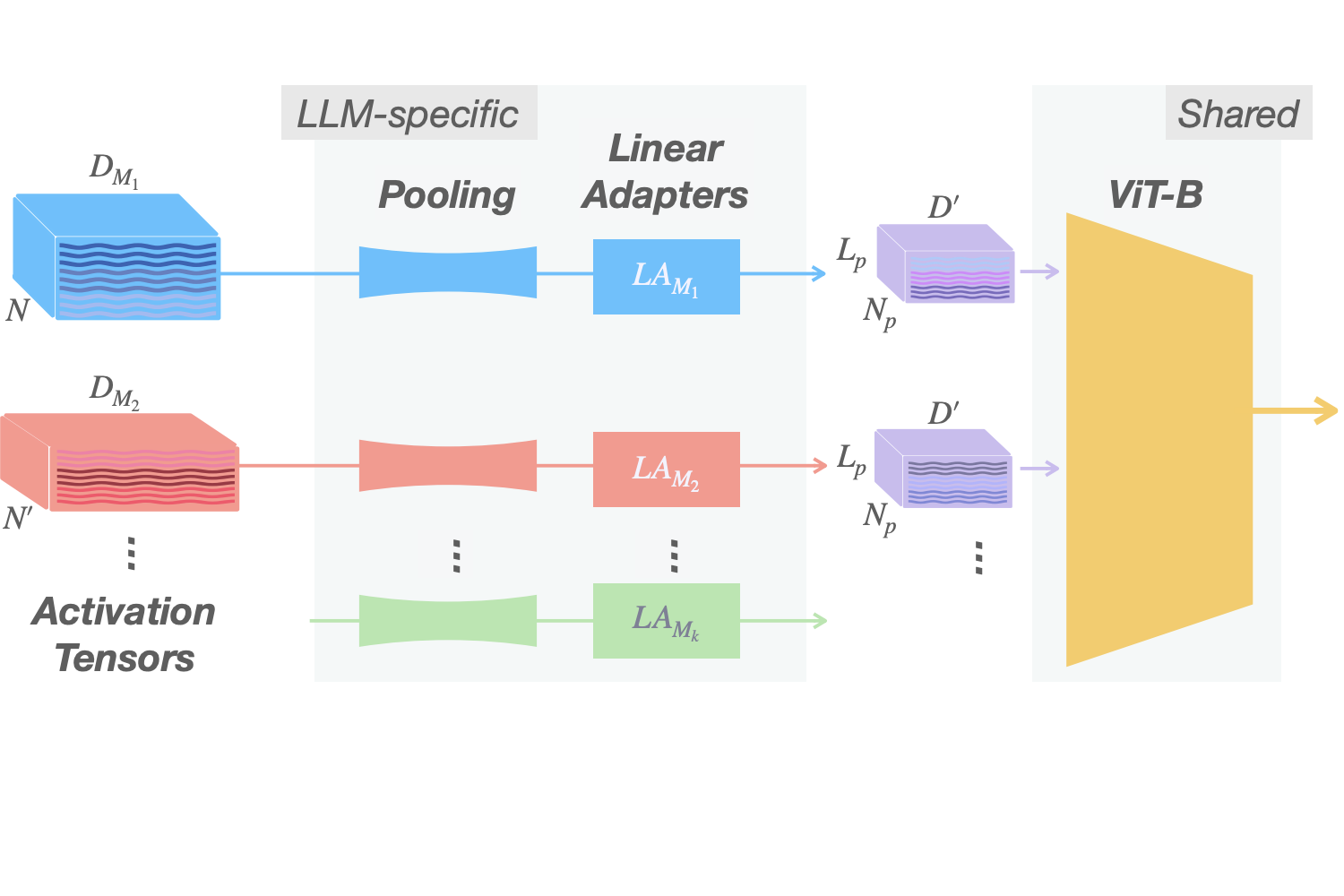
Beyond Token Probes: Hallucination Detection via Activation Tensors with ACT-ViT
Guy Bar-Shalom*, Fabrizio Frasca*, Yaniv Galron, Yftah Ziser, Haggai Maron
NeurIPS 2025
Abstract
Detecting hallucinations in Large Language Model-generated text is crucial for their safe deployment. While probing classifiers show promise, they operate on isolated layer–token pairs and are LLM-specific, limiting their effectiveness and hindering cross-LLM applications. In this paper, we introduce a novel approach to address these shortcomings. We build on the natural sequential structure of activation data in both axes (layers × tokens) and advocate treating full activation tensors akin to images. We design ACT-ViT, a Vision Transformer-inspired model that can be effectively and efficiently applied to activation tensors and supports training on data from multiple LLMs simultaneously. Through comprehensive experiments encompassing diverse LLMs and datasets, we demonstrate that ACT-ViT consistently outperforms traditional probing techniques while remaining extremely efficient for deployment. In particular, we show that our architecture benefits substantially from multi-LLM training, achieves strong zero-shot performance on unseen datasets, and can be transferred effectively to new LLMs through fine-tuning. Full code is available at https://github.com/BarSGuy/ACT-ViT.
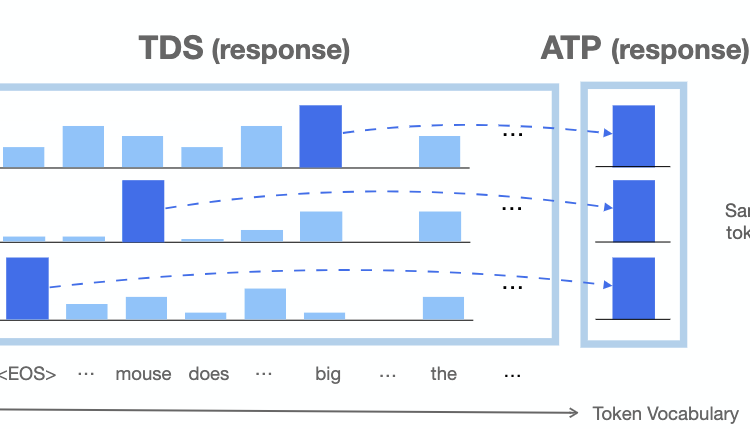
Beyond Next Token Probabilities: Learnable, Fast Detection of Hallucinations and Data Contamination on LLM Output Distributions
Guy Bar-Shalom*, Fabrizio Frasca*, Derek Lim, Yoav Gelberg, Yftah Ziser, Ran El-Yaniv, Gal Chechik, Haggai Maron
AAAI 2026
Abstract
The automated detection of hallucinations and training data contamination is pivotal to the safe deployment of Large Language Models (LLMs). These tasks are particularly challenging in settings where no access to model internals is available. Current approaches in this setup typically leverage only the probabilities of actual tokens in the text, relying on simple task-specific heuristics. Crucially, they overlook the information contained in the full sequence of next-token probability distributions. We propose to go beyond hand-crafted decision rules by learning directly from the complete observable output of LLMs -- consisting not only of next-token probabilities, but also the full sequence of next-token distributions. We refer to this as the LLM Output Signature (LOS), and treat it as a reference data type for detecting hallucinations and data contamination. To that end, we introduce LOS-Net, a lightweight attention-based architecture trained on an efficient encoding of the LOS, which can provably approximate a broad class of existing techniques for both tasks. Empirically, LOS-Net achieves superior performance across diverse benchmarks and LLMs, while maintaining extremely low detection latency. Furthermore, it demonstrates promising transfer capabilities across datasets and LLMs.
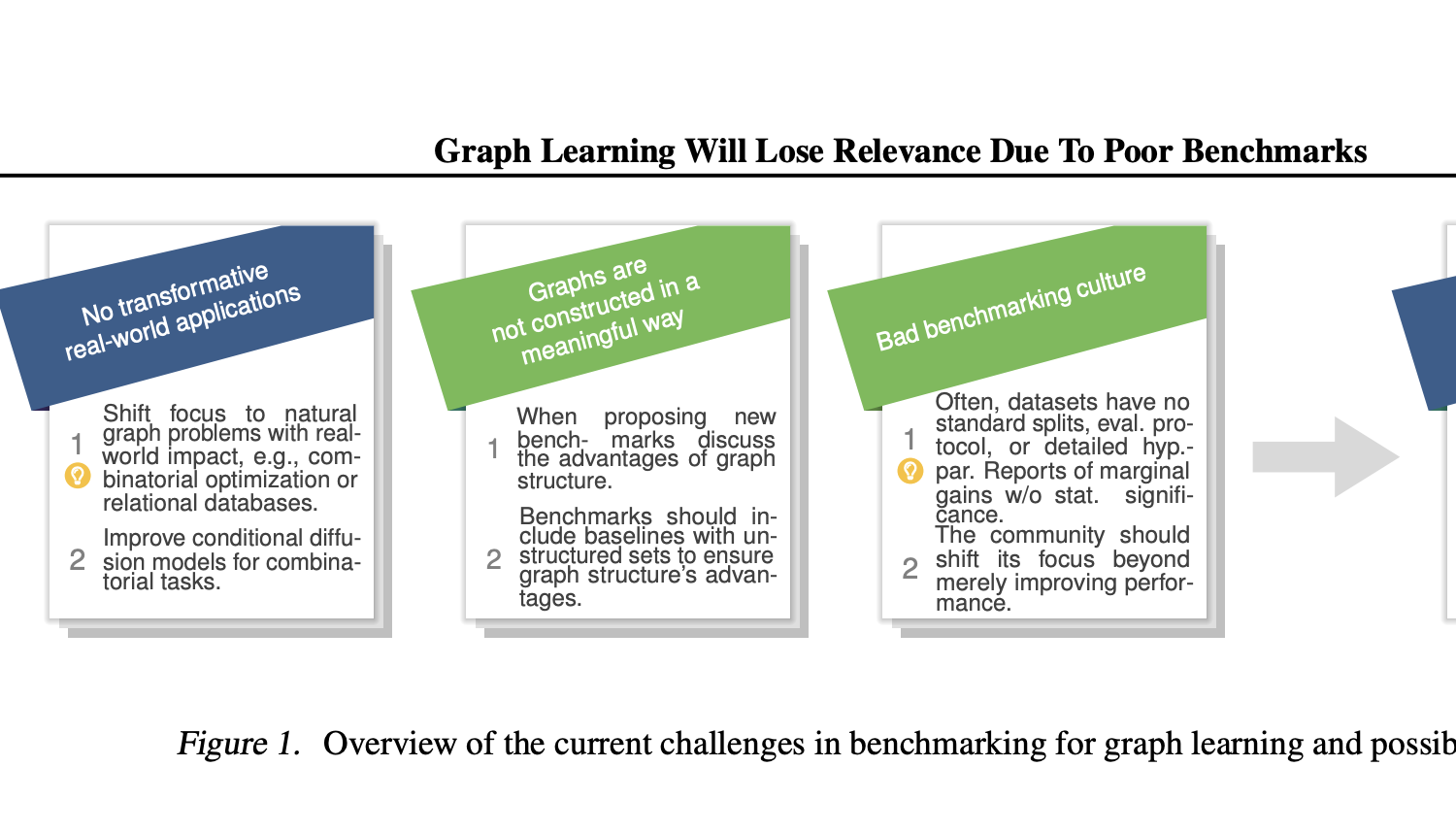
Position: Graph Learning Will Lose Relevance Due To Poor Benchmarks
Maya Bechler-Speicher*, Ben Finkelshtein*, Fabrizio Frasca*, Luis Müller*, Jan Tönshoff*, Antoine Siraudin, Viktor Zaverkin, Michael M. Bronstein, Mathias Niepert, Bryan Perozzi, Mikhail Galkin, Christopher Morris
(paper)
ICML 2025
Abstract
While machine learning on graphs has demonstrated promise in drug design and molecular property prediction, significant benchmarking challenges hinder its further progress and relevance. Current benchmarking practices often lack focus on transformative, real-world applications, favoring narrow domains like two-dimensional molecular graphs over broader, impactful areas such as combinatorial optimization, relational databases, or chip design. Additionally, many benchmark datasets poorly represent the underlying data, leading to inadequate abstractions and misaligned use cases. Fragmented evaluations and an excessive focus on accuracy further exacerbate these issues, incentivizing overfitting rather than fostering generalizable insights. These limitations have prevented the development of truly useful graph foundation models. This position paper calls for a paradigm shift toward more meaningful benchmarks, rigorous evaluation protocols, and stronger collaboration with domain experts to drive impactful and reliable advances in graph learning research, unlocking the potential of graph learning.
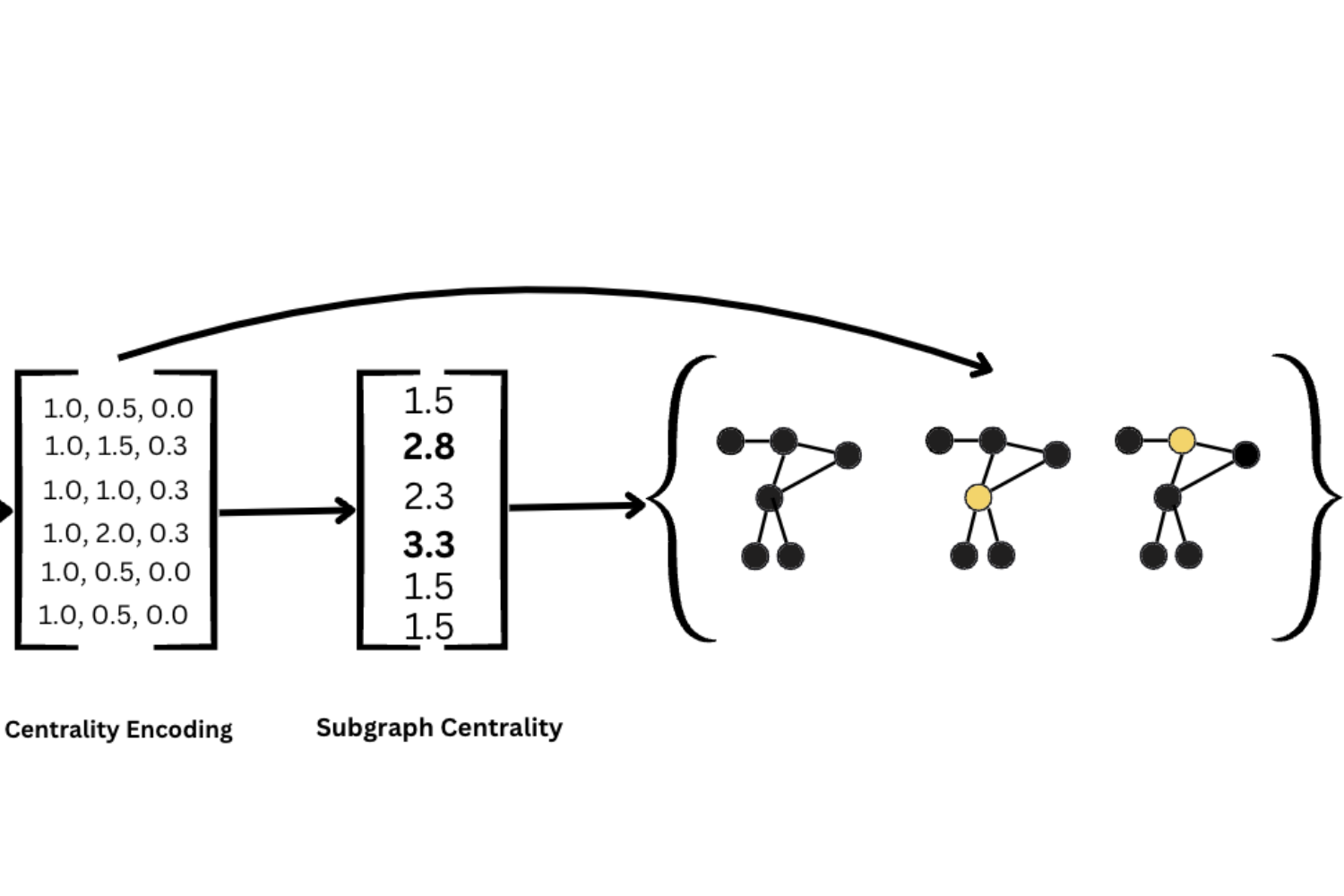
Balancing Efficiency and Expressiveness: Subgraph GNNs with Walk-Based Centrality
Joshua Southern*, Yam Eitan, Guy Bar-Shalom, Michael Bronstein, Haggai Maron, Fabrizio Frasca*
ICML 2025
Abstract
We propose an expressive and efficient approach that combines the strengths of two prominent extensions of Graph Neural Networks (GNNs): Subgraph GNNs and Structural Encodings (SEs). Our approach leverages walk-based centrality measures, both as a powerful form of SE and also as a subgraph selection strategy for Subgraph GNNs. By drawing a connection to perturbation analysis, we highlight the effectiveness of centrality-based sampling, and show it significantly reduces the computational burden associated with Subgraph GNNs. Further, we combine our efficient Subgraph GNN with SEs derived from the calculated centrality and demonstrate this hybrid approach, dubbed HyMN, gains in discriminative power. HyMN effectively addresses the expressiveness limitations of Message Passing Neural Networks (MPNNs) while mitigating the computational costs of Subgraph GNNs. Through a series of experiments on synthetic and real-world tasks, we show it outperforms other subgraph sampling approaches while being competitive with full-bag Subgraph GNNs and other state-of-the-art approaches with a notably reduced runtime.
Towards Foundation Models on Graphs: An Analysis on Cross-Dataset Transfer of Pretrained GNNs
Fabrizio Frasca, Fabian Jogl, Moshe Eliasof, Matan Ostrovsky, Carola-Bibiane Schönlieb, Thomas Gärtner, Haggai Maron
(paper)
Symmetry and Geometry in Neural Representations (NeurReps) @ NeurIPS 2024
Abstract
To develop a preliminary understanding towards Graph Foundation Models, we study the extent to which pretrained Graph Neural Networks can be applied across datasets, an effort requiring to be agnostic to dataset-specific features and their encodings. We build upon a purely structural pretraining approach and propose an extension to capture feature information while still being feature-agnostic. We evaluate pretrained models on downstream tasks for varying amounts of training samples and choices of pretraining datasets. Our preliminary results indicate that embeddings from pretrained models improve generalization only with enough downstream data points and in a degree which depends on the quantity and properties of pretraining data. Feature information can lead to improvements, but currently requires some similarities between pretraining and downstream feature spaces.
Understanding and Extending Subgraph GNNs by Rethinking Their Symmetries
Fabrizio Frasca*, Beatrice Bevilacqua*, Michael M. Bronstein, Haggai Maron
NeurIPS 2022
Oral (1.7% acceptance rate)
Abstract
Subgraph GNNs are a recent class of expressive Graph Neural Networks (GNNs) which model graphs as collections of subgraphs. So far, the design space of possible Subgraph GNN architectures as well as their basic theoretical properties are still largely unexplored. In this paper, we study the most prominent form of subgraph methods, which employs node-based subgraph selection policies such as ego-networks or node marking and deletion. We address two central questions: (1) What is the upper-bound of the expressive power of these methods? and (2) What is the family of equivariant message passing layers on these sets of subgraphs?. Our first step in answering these questions is a novel symmetry analysis which shows that modelling the symmetries of node-based subgraph collections requires a significantly smaller symmetry group than the one adopted in previous works. This analysis is then used to establish a link between Subgraph GNNs and Invariant Graph Networks (IGNs). We answer the questions above by first bounding the expressive power of subgraph methods by 3-WL, and then proposing a general family of message-passing layers for subgraph methods that generalises all previous node-based Subgraph GNNs. Finally, we design a novel Subgraph GNN dubbed SUN, which theoretically unifies previous architectures while providing better empirical performance on multiple benchmarks.
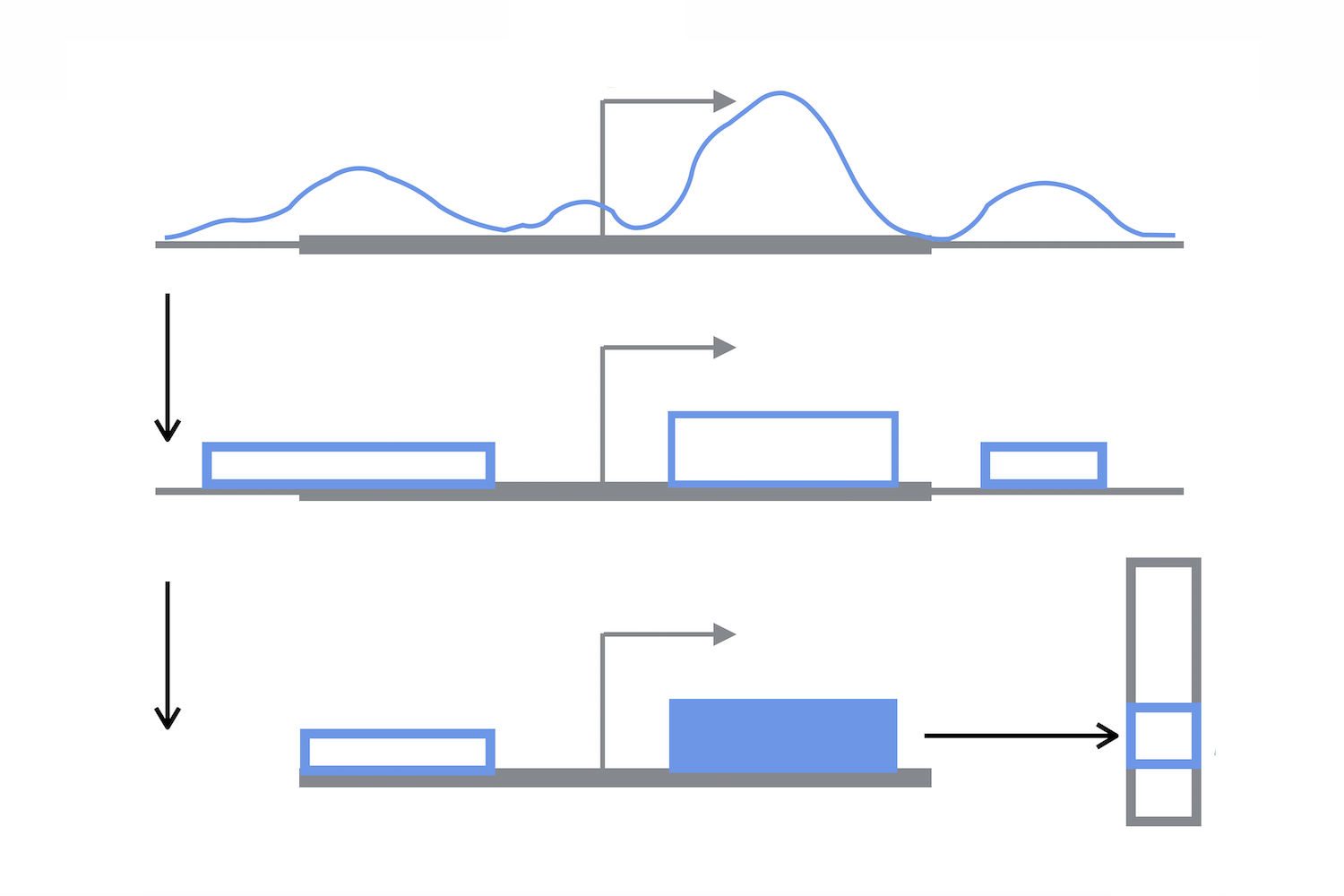
Accurate and Highly Interpretable Prediction of Gene Expression from Histone Modifications
Fabrizio Frasca, Matteo Matteucci, Michele Leone, Marco J. Morelli, Marco Masseroli
BMC Bioinformatics (2022)
Abstract
Histone Mark Modifications (HMs) are crucial actors in gene regulation, as they actively remodel chromatin to modulate transcriptional activity: aberrant combinatorial patterns of HMs have been connected with several diseases, including cancer. HMs are, however, reversible modifications: understanding their role in disease would allow the design of ‘epigenetic drugs’ for specific, non-invasive treatments. Standard statistical techniques were not entirely successful in extracting representative features from raw HM signals over gene locations. On the other hand, deep learning approaches allow for effective automatic feature extraction, but at the expense of model interpretation. Here, we propose ShallowChrome, a novel computational pipeline to model transcriptional regulation via HMs in both an accurate and interpretable way. We attain state-of-the-art results on the binary classification of gene transcriptional states over 56 cell-types from the REMC database, largely outperforming recent deep learning approaches. We interpret our models by extracting insightful gene-specific regulative patterns, and we analyse them for the specific case of the PAX5 gene over three differentiated blood cell lines. Finally, we compare the patterns we obtained with the characteristic emission patterns of ChromHMM, and show that ShallowChrome is able to coherently rank groups of chromatin states w.r.t. their transcriptional activity. In this work we demonstrate that it is possible to model HM-modulated gene expression regulation in a highly accurate, yet interpretable way. Our feature extraction algorithm leverages on data downstream the identification of enriched regions to retrieve gene-wise, statistically significant and dynamically located features for each HM. These features are highly predictive of gene transcriptional state, and allow for accurate modeling by computationally efficient logistic regression models. These models allow a direct inspection and a rigorous interpretation, helping to formulate quantifiable hypotheses.
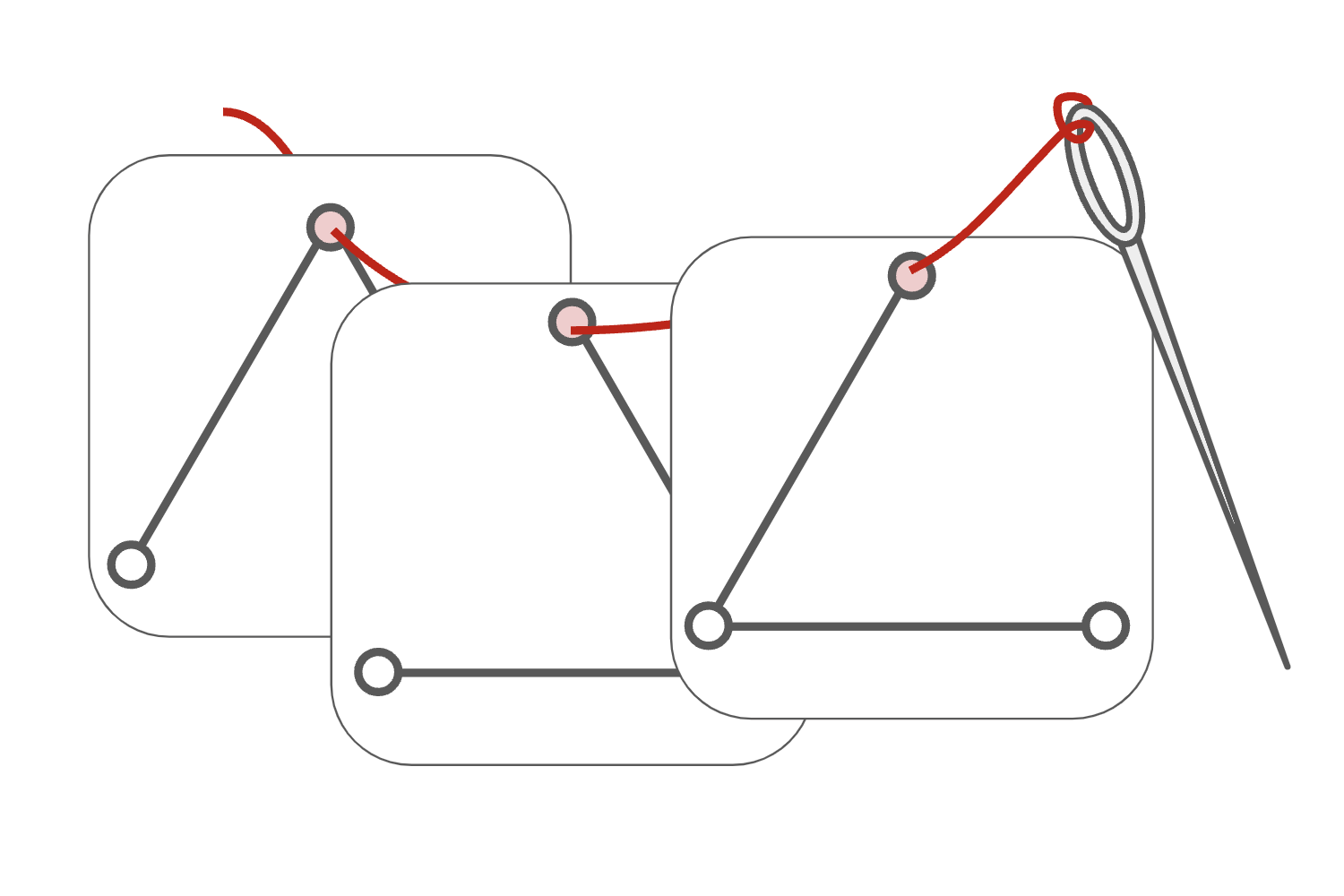
Equivariant Subgraph Aggregation Networks
Beatrice Bevilacqua*, Fabrizio Frasca*, Derek Lim*, Balasubramaniam Srinivasan, Chen Cai, Gopinath Balamurugan, Michael M. Bronstein, Haggai Maron
ICLR 2022
Spotlight (5% acceptance rate)
Abstract
Message-passing neural networks (MPNNs) are the leading architecture for deep learning on graph-structured data, in large part due to their simplicity and scalability. Unfortunately, it was shown that these architectures are limited in their expressive power. This paper proposes a novel framework called Equivariant Subgraph Aggregation Networks (ESAN) to address this issue. Our main observation is that while two graphs may not be distinguishable by an MPNN, they often contain distinguishable subgraphs. Thus, we propose to represent each graph as a set of subgraphs derived by some predefined policy, and to process it using a suitable equivariant architecture. We develop novel variants of the 1-dimensional Weisfeiler-Leman (1-WL) test for graph isomorphism, and prove lower bounds on the expressiveness of ESAN in terms of these new WL variants. We further prove that our approach increases the expressive power of both MPNNs and more expressive architectures. Moreover, we provide theoretical results that describe how design choices such as the subgraph selection policy and equivariant neural architecture affect our architecture's expressive power. To deal with the increased computational cost, we propose a subgraph sampling scheme, which can be viewed as a stochastic version of our framework. A comprehensive set of experiments on real and synthetic datasets demonstrates that our framework improves the expressive power and overall performance of popular GNN architectures.
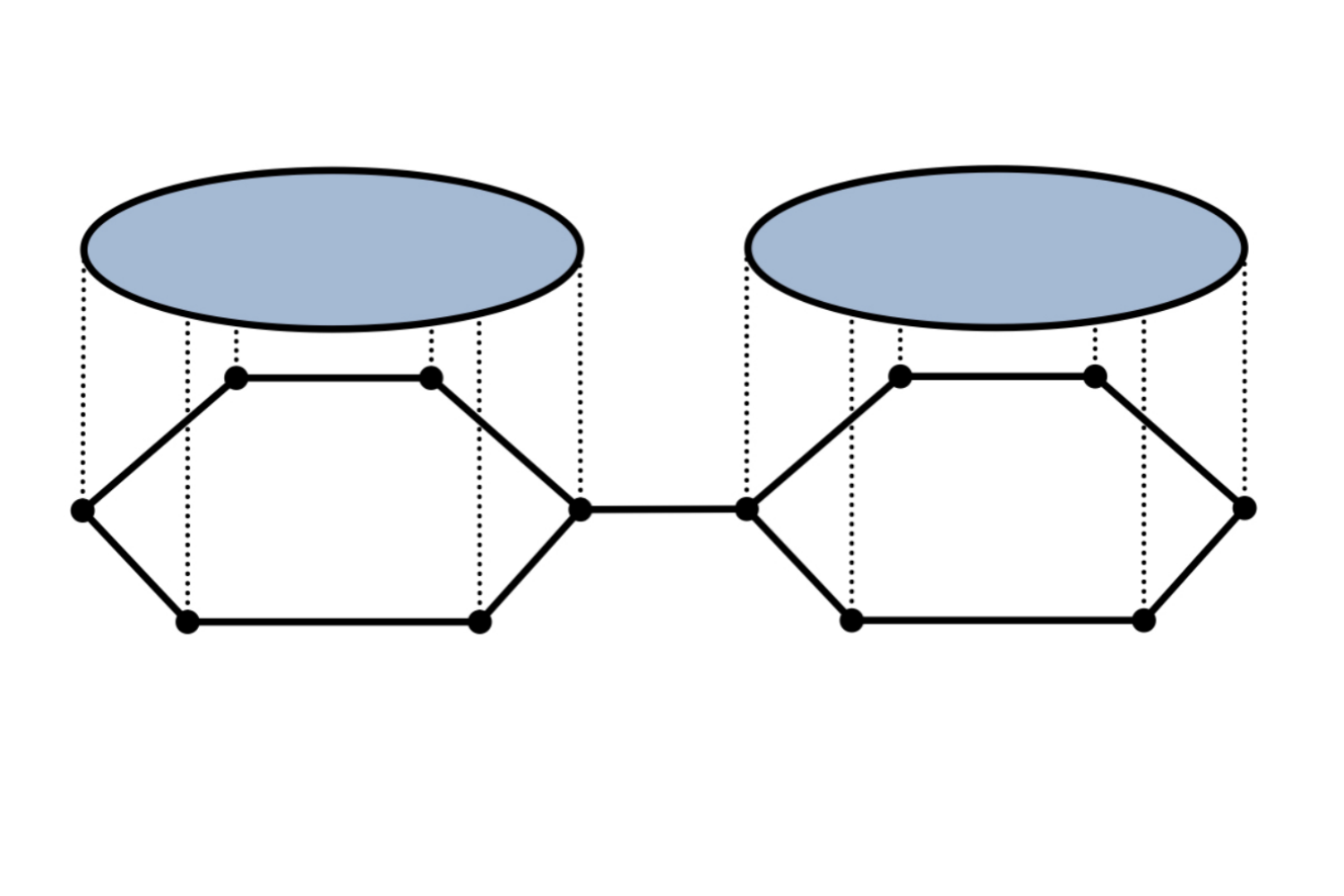
Weisfeiler and Lehman Go Cellular: CW Networks
Cristian Bodnar*, Fabrizio Frasca*, Nina Otter, Yu Guang Wang, Pietro Liò, Guido Montúfar, Michael M. Bronstein
NeurIPS 2021
Abstract
Graph Neural Networks (GNNs) are limited in their expressive power, struggle with long-range interactions and lack a principled way to model higher-order structures. These problems can be attributed to the strong coupling between the computational graph and the input graph structure. The recently proposed Message Passing Simplicial Networks naturally decouple these elements by performing message passing on the clique complex of the graph. Nevertheless, these models can be severely constrained by the rigid combinatorial structure of Simplicial Complexes (SCs). In this work, we extend recent theoretical results on SCs to regular Cell Complexes, topological objects that flexibly subsume SCs and graphs. We show that this generalisation provides a powerful set of graph "lifting" transformations, each leading to a unique hierarchical message passing procedure. The resulting methods, which we collectively call CW Networks (CWNs), are strictly more powerful than the WL test and not less powerful than the 3-WL test. In particular, we demonstrate the effectiveness of one such scheme, based on rings, when applied to molecular graph problems. The proposed architecture benefits from provably larger expressivity than commonly used GNNs, principled modelling of higher-order signals and from compressing the distances between nodes. We demonstrate that our model achieves state-of-the-art results on a variety of molecular datasets.
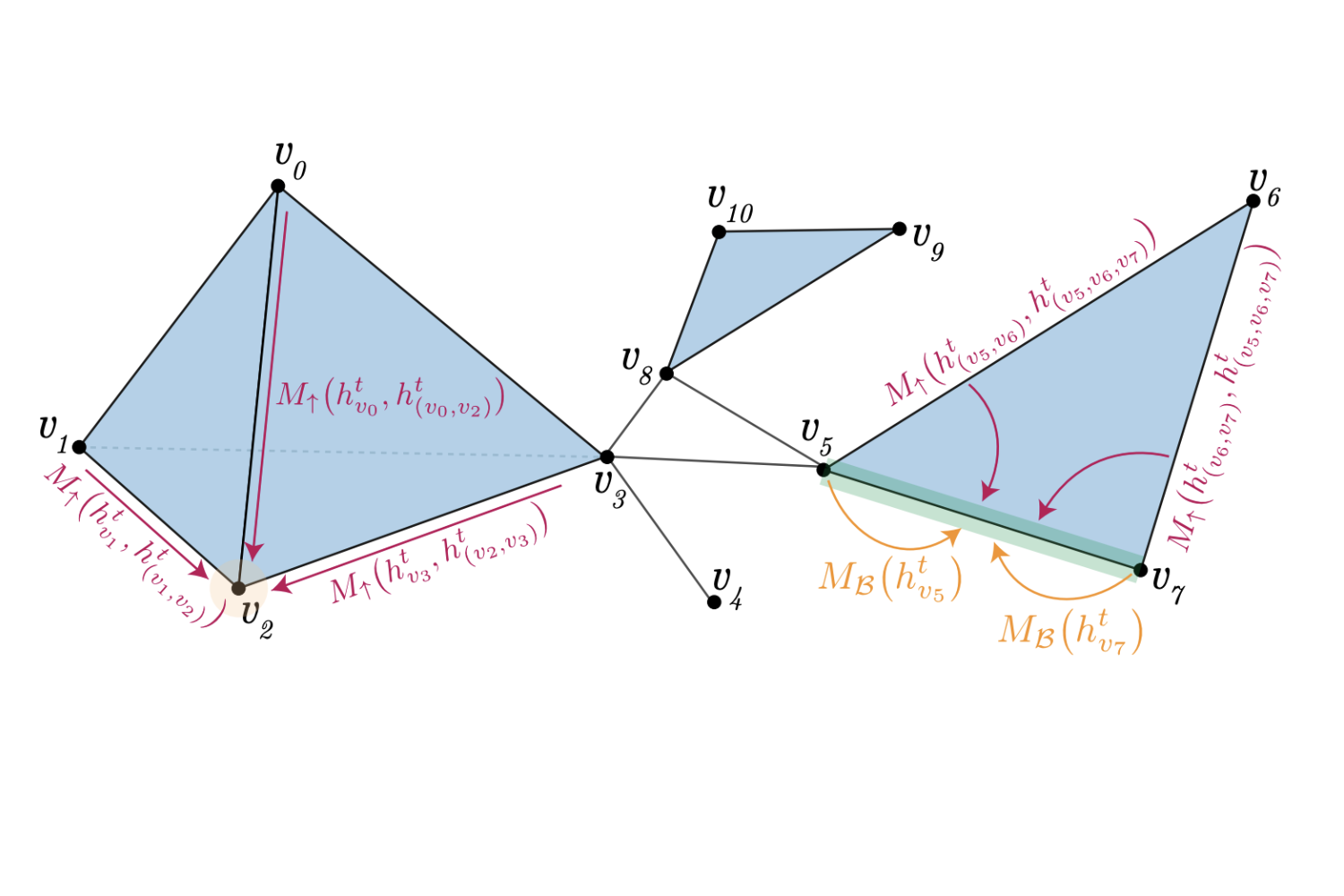
Weisfeiler and Lehman Go Topological: Message Passing Simplicial Networks
Cristian Bodnar*, Fabrizio Frasca*, Yu Guang Wang*, Nina Otter, Guido Montúfar*, Pietro Liò, Michael M. Bronstein
ICML 2021
Abstract
The pairwise interaction paradigm of graph machine learning has predominantly governed the modelling of relational systems. However, graphs alone cannot capture the multi-level interactions present in many complex systems and the expressive power of such schemes was proven to be limited. To overcome these limitations, we propose Message Passing Simplicial Networks (MPSNs), a class of models that perform message passing on simplicial complexes (SCs). To theoretically analyse the expressivity of our model we introduce a Simplicial Weisfeiler-Lehman (SWL) colouring procedure for distinguishing non-isomorphic SCs. We relate the power of SWL to the problem of distinguishing non-isomorphic graphs and show that SWL and MPSNs are strictly more powerful than the WL test and not less powerful than the 3-WL test. We deepen the analysis by comparing our model with traditional graph neural networks (GNNs) with ReLU activations in terms of the number of linear regions of the functions they can represent. We empirically support our theoretical claims by showing that MPSNs can distinguish challenging strongly regular graphs for which GNNs fail and, when equipped with orientation equivariant layers, they can improve classification accuracy in oriented SCs compared to a GNN baseline.
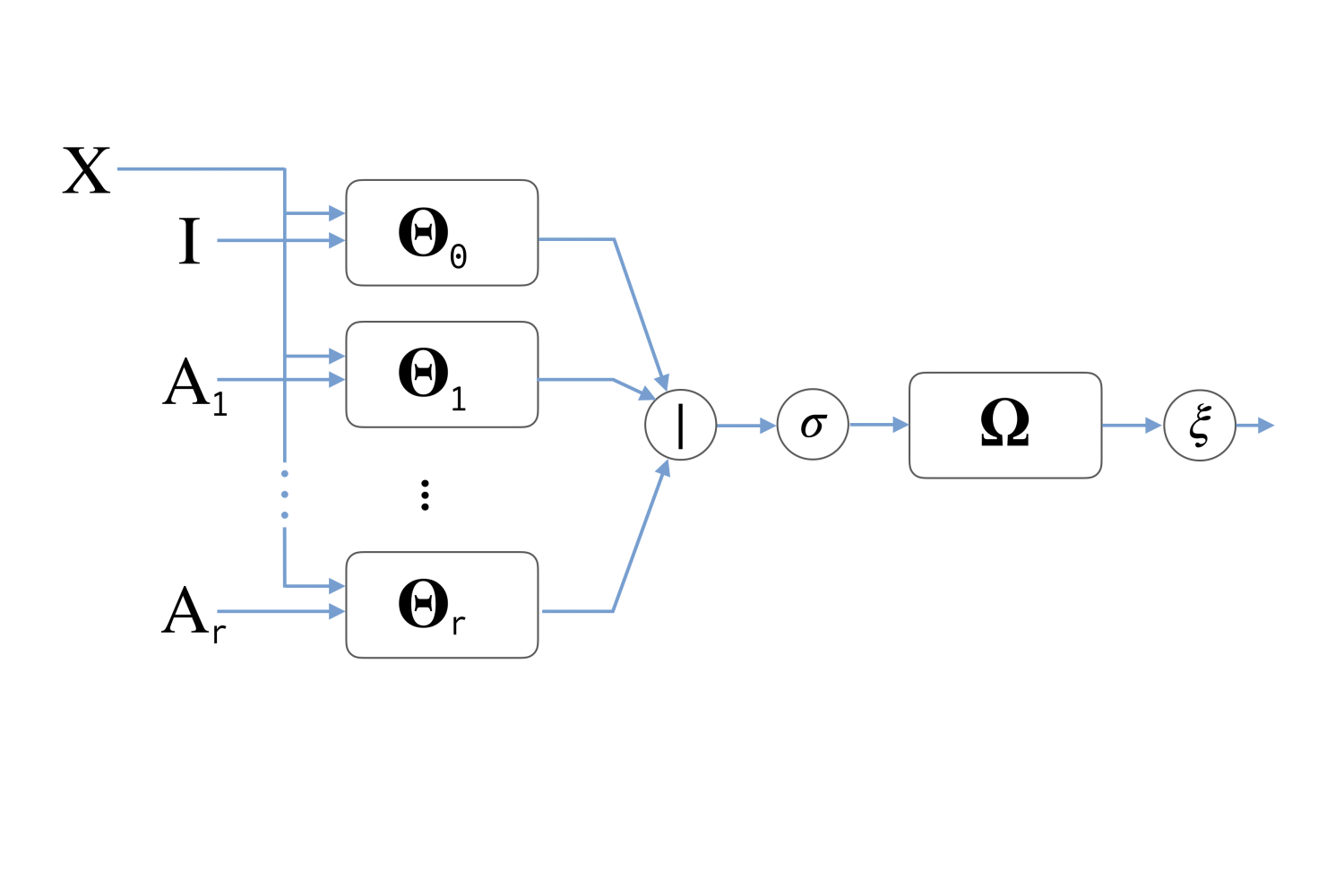
Scalable Inception Graph Neural Networks
Fabrizio Frasca*, Emanuele Rossi*, Davide Eynard, Ben Chamberlain, Michael M. Bronstein, Federico Monti
Abstract
Graph representation learning has recently been applied to a broad spectrum of problems ranging from computer graphics and chemistry to high energy physics and social media. The popularity of graph neural networks has sparked interest, both in academia and in industry, in developing methods that scale to very large graphs such as Facebook or Twitter social networks. In most of these approaches, the computational cost is alleviated by a sampling strategy retaining a subset of node neighbors or subgraphs at training time. In this paper we propose a new, efficient and scalable graph deep learning architecture which sidesteps the need for graph sampling by using graph convolutional filters of different size that are amenable to efficient precomputation, allowing extremely fast training and inference. Our architecture allows using different local graph operators (e.g. motif-induced adjacency matrices or Personalized Page Rank diffusion matrix) to best suit the task at hand. We conduct extensive experimental evaluation on various open benchmarks and show that our approach is competitive with other state-of-the-art architectures, while requiring a fraction of the training and inference time. Moreover, we obtain state-of-the-art results on ogbn-papers100M, the largest public graph dataset, with over 110 million nodes and 1.5 billion edges.
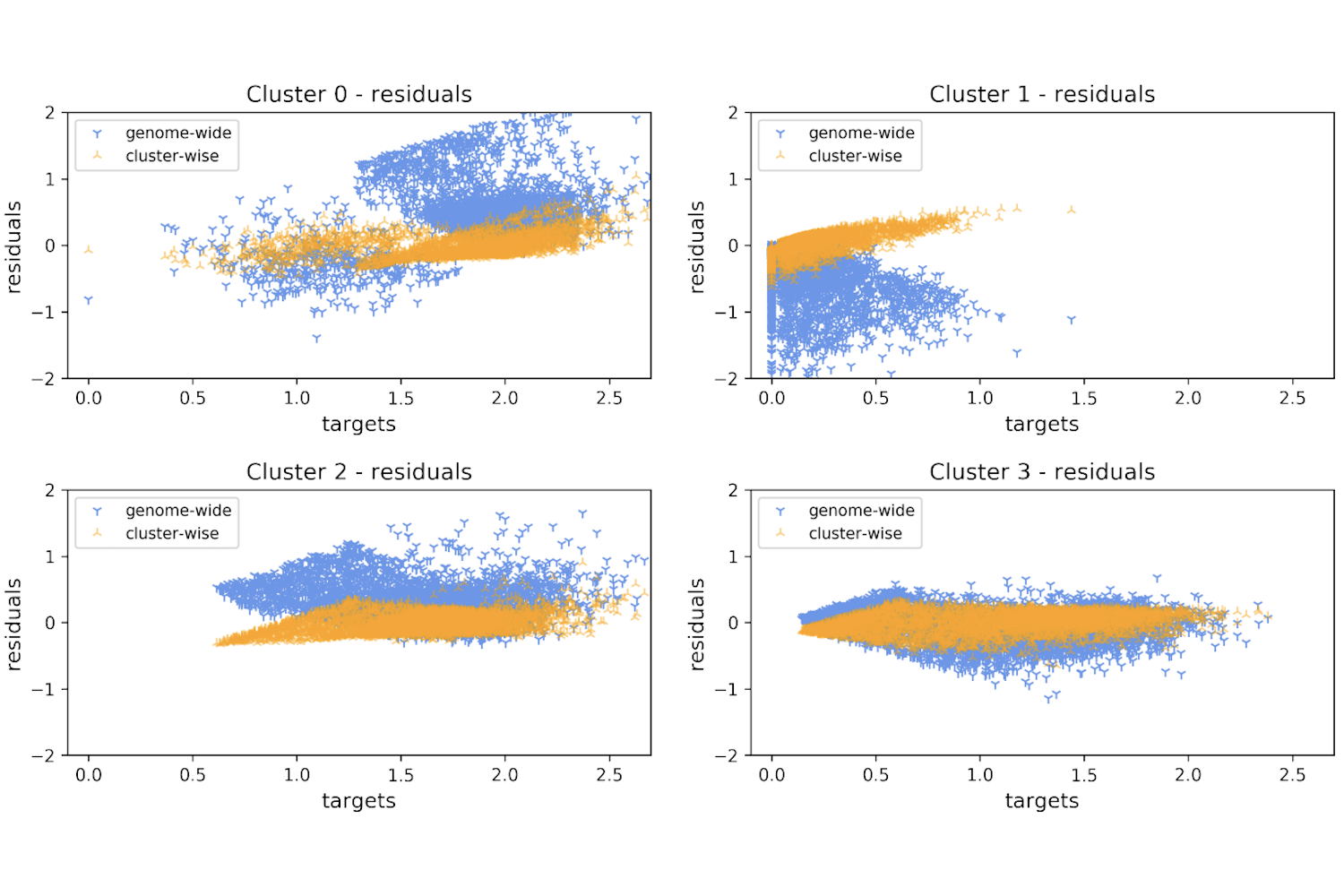
Exposing and Characterizing Subpopulations of Distinctly Regulated Genes by K-Plane Regression
Fabrizio Frasca, Matteo Matteucci, Marco J. Morelli, Marco Masseroli
(paper)
LNBI (Lecture Notes in BioInformatics, 2020; extended from CIBB 2018)
Abstract
Understanding the roles and interplays of histone marks and transcription factors in the regulation of gene expression is of great interest in the development of non-invasive and personalized therapies. Computational studies at genome-wide scale represent a powerful explorative framework, allowing to draw general conclusions. However, a genome-wide approach only identifies generic regulative motifs, and possible multi-functional or co-regulative interactions may remain concealed. In this work, we hypothesize the presence of a number of distinct subpopulations of transcriptional regulative patterns within the set of protein coding genes that explain the statistical redundancy observed at a genome-wide level. We propose the application of a K-Plane Regression algorithm to partition the set of protein coding genes into clusters with specific shared regulative mechanisms. Our approach is completely data-driven and computes clusters of genes significantly better fitted by specific linear models, in contrast to single regressions. These clusters are characterized by distinct and sharper histonic input patterns, and different mean expression values.
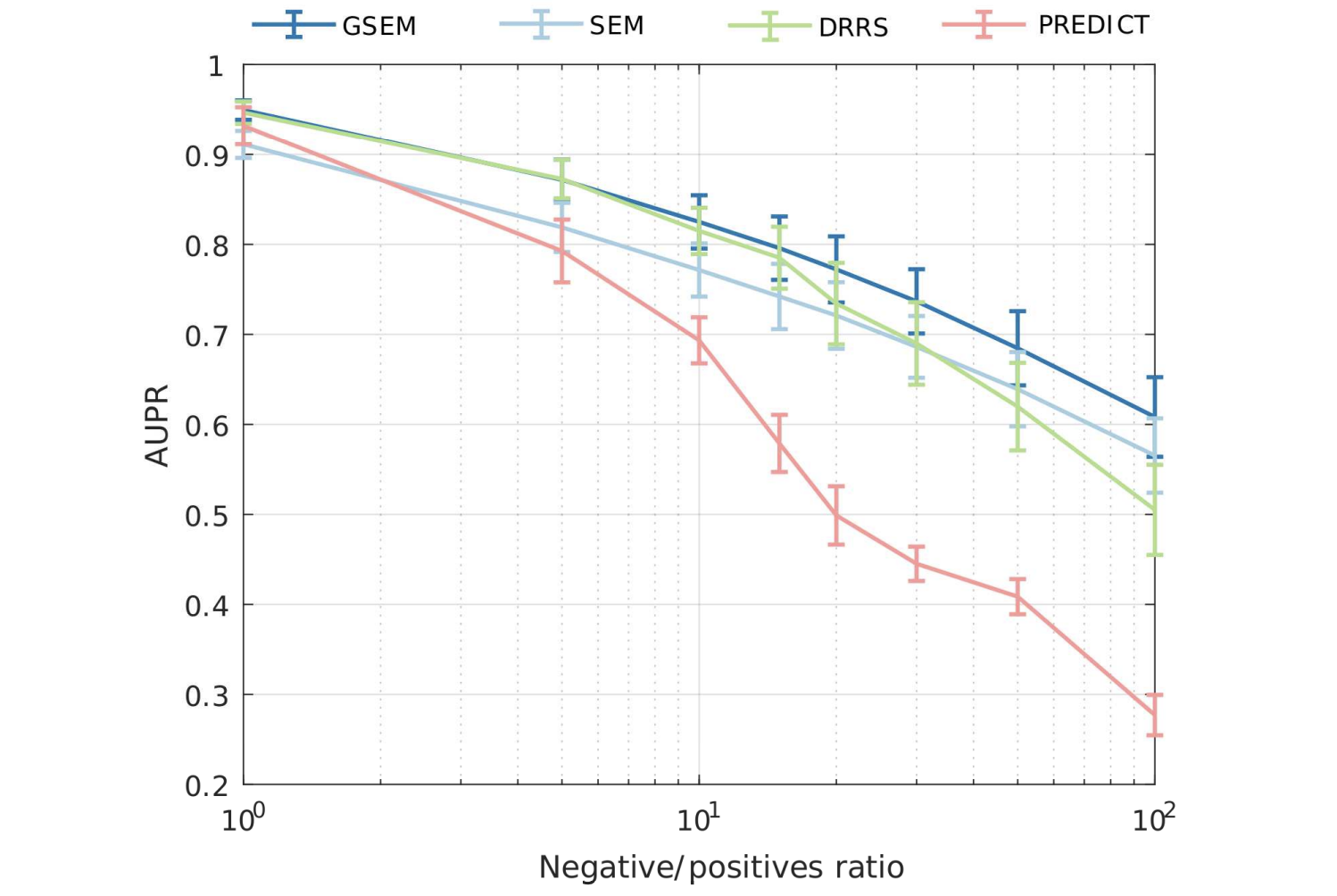
Learning Interpretable Disease Self-Representations for Drug Repositioning
Fabrizio Frasca*, Diego Galeano*, Guadalupe Gonzalez, Ivan Laponogov, Kirill Veselkov, Alberto Paccanaro, Michael M. Bronstein
Abstract
Drug repositioning is an attractive cost-efficient strategy for the development of treatments for human diseases. Here, we propose an interpretable model that learns disease self-representations for drug repositioning. Our self-representation model represents each disease as a linear combination of a few other diseases. We enforce proximity in the learnt representations in a way to preserve the geometric structure of the human phenome network - a domain-specific knowledge that naturally adds relational inductive bias to the disease self-representations. We prove that our method is globally optimal and show results outperforming state-of-the-art drug repositioning approaches. We further show that the disease self-representations are biologically interpretable.
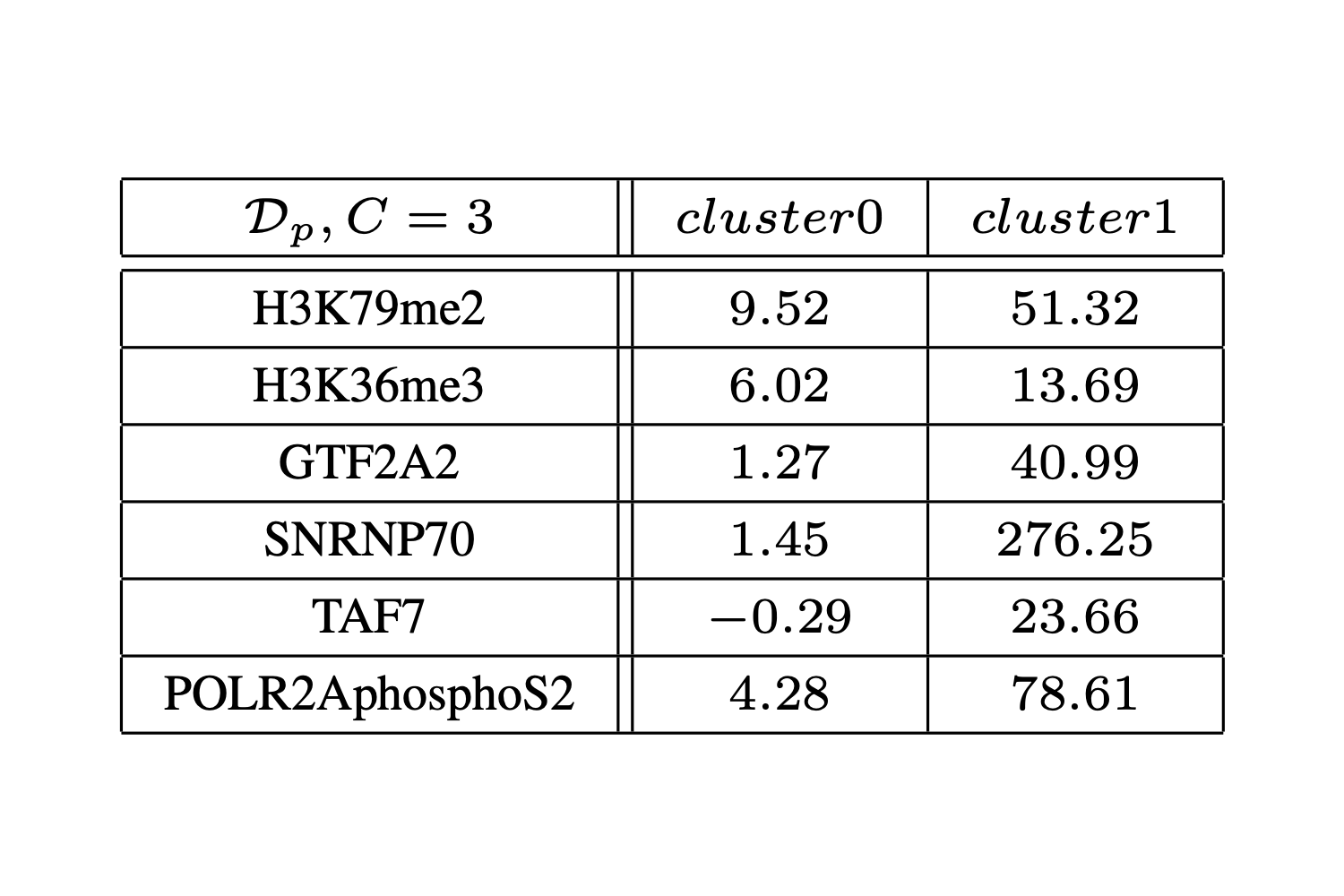
Modeling Gene Transcriptional Regulation by Means of Hyperplanes Genetic Clustering
Fabrizio Frasca, Matteo Matteucci, Marco Masseroli, Marco J. Morelli
(paper)
IJCNN 2018
Abstract
In the wide context of biological processes regulating gene expression, transcriptional regulation driven by epigenetic activity is among the most effective and intriguing ones. Understanding the complex language of histone modifications and transcription factor bindings is an appealing yet hard task, given the large number of involved features and the specificity of their combinatorial behavior across genes. Genome-wide regression models for predicting mRNA abundance quantifications from epigenetic activity are interesting in an exploratory framework, but their effectiveness is limited as the relative predictive power of epigenetic features is hard to discern at such level of resolution. On the other hand, an investigative analysis cannot rely on prior biological knowledge to perform sensible grouping of genes and locally study epigenetic regulative processes. In this context, we shaped the “gene stratification problem” as a form of epigenetic feature-based hyperplanes clustering, and proposed a genetic algorithm to approach this task, aiming at performing datadriven partitioning of the whole set of protein coding genes of an organism based on the characteristic relation between their expression and the associated epigenetic activity. We observed how, not only the hyperplanes described by the resulting partitions significantly differ from each other, but also how different epigenetic features are of diverse importance in predicting gene expression within each partition. This demonstrates the validity and biological interest of the proposed computational method and the obtained results.
My personal website has been realised through Jekyll and Github Pages. The theme — slightly customised — is by orderedlist. Drop me a message if you'd fancy a chat!